
Il deficit di Alfa1 Antitripsina (DAAT) è una condizione genetica ereditaria rara. Gli individui con deficit di AAT presentano fin da bambini livelli ridotti o addirittura assenti di tale proteina. Essa, in situazioni normali, viene prodotta dal fegato per poi essere riversata nel sangue e la sua principale funzione è quella di proteggere i polmoni dai vari fenomeni infiammatori che li possono interessare (come quelli che accompagnano infezioni oppure quelli indotti da sostanze chimiche irritanti). Questa condizione ereditaria è associata principalmente all’insorgenza di malattie dei polmoni e/o malattie del fegato, ma anche altre condizioni come pannicolite e vasculite.
Causa del deficit di AAT
Per condizione genetica si intende una qualsiasi alterazione, patologica o innocua, la cui causa è situata nel DNA, il quale include tutte le informazioni per la produzione delle proteine dell’organismo, compresa AAT. Tali informazioni sono organizzate in geni (vale quindi, in generale, l’equivalenza un gene-una proteina) ed in ciascun individuo sono presenti due geni, uno di origine materna ed uno di origina paterna. Questi, insieme, permettono la produzione della proteina in corrette quantità. I due geni che vengono a trovarsi nel medesimo individuo possono differire tra loro e prendono il nome di varianti alleliche (o alleli).
Il gene che trasporta le informazioni per produrre AAT (il cui nome è SERPINA1) può presentarsi in una variante allelica normale (M) che permette la produzione di adeguate quantità di proteina. Esistono, però, anche varianti alleliche alterate (come S, Z, Null, Malton…) che invece permettono una produzione in quantità anomale, inferiori o nulle, della proteina.
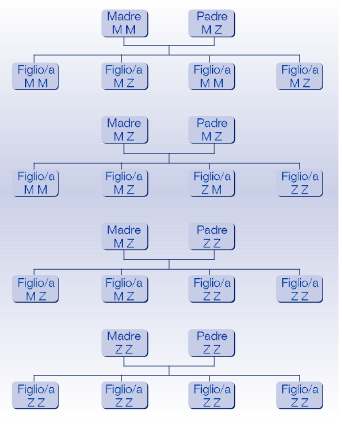
Essendo presenti due alleli per la stessa proteina in ogni individuo, le combinazioni possono essere differenti e ciascuna è associata ad una diversa quantità di AAT prodotta (gli alleli vengono infatti espressi in maniera co-dominante). Una diminuzione di tale quantità non porta necessariamente alla comparsa di malattia, ma determina una predisposizione allo sviluppo di alcune patologie correlate. Ecco alcuni esempi:
- MM – Individuo con normali livelli di AAT
- MZ – Individuo eterozigote con deficit intermedio di AAT e rischio di sviluppare patologie correlate lievemente aumentato
- ZZ – Individuo omozigote con deficit severo di AAT e rischio di sviluppare patologie correlate
- SZ, ZI, ZMalton ecc. – Individuo eterozigote composto con deficit severo e rischio di sviluppare patologie correlate
Un individuo può trasmettere una delle sue due varianti alleliche alla prole e per questo motivo il deficit di AAT è definito come condizione ereditaria. La possibilità che un figlio presenti il deficit è determinata dalle varianti alleliche che i due genitori possono tramandare. Un esempio di trasmissione è riportato nell’immagine.
Manifestazioni del deficit di AAT
Sulla base delle informazioni contenute dai geni, la proteina AAT viene prodotta dalle cellule del fegato e riversata nel sangue. Quest’ultimo provvede al trasporto della proteina ai tessuti periferici e specialmente ai polmoni, in cui AAT svolge la sua principale azione. Fegato e polmoni sono quindi strettamente legati alla proteina Alfa1 Antitripsina ed infatti sono gli organi maggiormente interessati dalle malattie associate al deficit.
A livello polmonare, le patologie principali associate al DAAT sono enfisema polmonare giovanile, BPCO (Bronco-Pneumopatia Cronica Ostruttiva), bronchiectasie, asma, pneumotorace. Lo sviluppo di enfisema generalmente procede in maniera subclinica per lunghi periodi, ma nelle fasi avanzate è associato ai seguenti sintomi:
- Respiro affannoso
- Sibilo durante il respiro
- Tosse cronica con produzione di catarro
Nel fegato, il deficit può causare epatopatia che compare già nel bambino ma che spesso va incontro a risoluzione. Talvolta, nel tempo, essa può evolvere a cirrosi durante la vita adulta. I principali sintomi sono:
- Ittero dovuto ad epatopatia (nei bambini), cioè assunzione da parte di occhi e cute di una colorazione gialla.
- Ascite (negli adulti), cioè gonfiore addominali associato alle fasi più avanzate della malattia, cioè alla cirrosi
Oltre a fegato e polmoni, seppur raramente, il DAAT è associato anche all’insorgenza di patologie a carico di altri organi come il sottocute (nel caso di pannicolite) e i vasi sanguigni (nel caso di vasculiti).
Il deficit di AAT è comunque caratterizzato da estrema variabilità per quanto riguarda le manifestazioni cliniche, che vanno dalle più semplici alle più severe. Questa variabilità dipende in primo luogo da quali varianti alleliche alterate determinano il deficit, ma anche dall’eventuale presenza di fattori di rischio che favoriscono la comparsa delle patologie associate.
Diffusione del deficit di AAT
Studi epidemiologici suggeriscono che il deficit colpisca 1 individuo su 2000-5000 abitanti, ma questi valori variano con la localizzazione geografica presa in considerazione: si riscontra un gradiente di prevalenza decrescente procedendo da nord a sud. La prevalenza, infatti, è massima nelle regioni del Nord Europa e decresce procedendo nelle regioni più meridionali.
Più che una condizione rara, la DAAT è una condizione raramente diagnosticata ed è ragionevole pensare che la prevalenza sia in realtà maggiore. Nella maggior parte delle persone con deficit, infatti, la diagnosi è imprecisa o del tutto assente a causa di tre principali motivi: essa non conduce necessariamente a malattia; i sintomi variano notevolmente da paziente a paziente (sia quantitativamente sia qualitativamente); altre condizioni sono associate alla medesima sintomatologia, esponendo al rischio di scorretta diagnosi.
IL DEFICIT DI ALFA 1AT
Cos’è e in cosa consiste
Il Deficit Alfa-1 Antitripsina è una condizione genetica rara caratterizzata da livelli bassi o nulli di tale proteina nell’organismo.
IL DEFICIT DI ALFA 1AT
Come curarlo
La cura del deficit alfa 1 antitripsina prevede delle terapie per la malattia polmonare ed epatica e l’ossigenoterapia.

IL DEFICIT DI ALFA 1AT
Come Diagnosticarlo
Alcune semplici informazioni su come diagnosticare il Deficit Alfa-1 Antitripsina, chi deve fare il test e cosa fare se l’esito è positivo.

IL DEFICIT DI ALFA 1AT
Come curarlo
La cura del deficit alfa 1 antitripsina prevede delle terapie per la malattia polmonare ed epatica e l’ossigenoterapia.
IL DEFICIT DI ALFA 1AT
Vivere con il deficit
Vivere con il deficit di Alfa-1 AT richiede una riabilitazione respiratoria, l’eliminazione del fumo e un’alimentazione corretta.
